Biography
Interests
Maria Carmen Agnello
Lawyer at the Hospital Management Purchasing Unit, Free Professional Collaboration with the Department of Legal Sciences of the University of Bologna
*Correspondence to: Dr. Maria Carmen Agnello, Lawyer at the Hospital Management Purchasing Unit, Free Professional Collaboration with the Department of Legal Sciences of the University of Bologna.
Contact: ARNAS Garibaldi in Catania, Email: agnelloc4@gmail.com
Copyright © 2019 Dr. Maria Carmen Agnello. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
The article analyses the set of measures put in place at different levels of regulation and programming to ensure a fair distribution of orphan drugs compared to the increasing diversification of rare diseases. These measures are preventive in supporting research and production in abandoned pharmaceutical sectors, as they are not profitable. At the same time, safety measures had to be taken not only to monitor but also to assess the costs and benefits. A specific focus is dedicated to the different distribution of the pay back provided by the last Finance Act of 2019 in order to encourage research and production in this sector.
Index
1.The Socio-legal Impact of Rare Diseases and Growth Data
2. The Regulatory and Programming Framework
2.1 The Context of the European Single Market
2.2 The Context of the National Health System
2.3 The Implications of the Financial Law for orphan drugs: the pay back measure
3. Actions and Measures for Sustainability
3.1 The Role of the AIFA: Supervision and Financing Medicine
3.2 Orphan Drugs and Off-Label Prescriptions
4. Concluding Aspects: The Social Welfare Impact, Comparison Between Economic and
Organizational Measures and Balancing Instruments
1. The Socio-legal Impact of Rare Diseases and Growth Data
The definition of orphan drugs is linked to diseases defined as “rare”, because they are administered to hyper
pathologies with a limited number of people affected [1].
The first issue concerns the production of such medicinal products, for the following reasons. In the past, pharmaceutical companies have had no interest in investing in this sector, which is targeted at a limited part of the market, compared to other diseases. Production and distribution was not profitable as it did not allow for the recovery of investment in research and production costs. This led to the definition of orphan drugs, which were abandoned by manufacturers, who preferred to focus research on high-density diseases.
This situation has changed due to the increasing prevalence of rare diseases, estimated at more than 30 million in Europe, while in Italy 2-3 million cases, with a consequent increase in the socioeconomic impact in terms of increasing demand for such drugs. This increase in demand corresponds to a slower response in supply due to high production and distribution costs [2].
This is related to the challenge of the sustainability of health systems through organizational and management solutions that can improve the levels of appropriateness in the administration of such drugs. The report of Pharmaceutical Research and Manufacturers of America indicate the development of 566 drugs, of which 233 for rare tumours; 148 for genetic disorders; 38 for neurological diseases; 31 for infectious diseases; 25 for autoimmune diseases. In Europe orphan drug designations have tripled from 80 in 2006 to 209 in 2016. A survey of farmindustria and Bain & Company has predicted in the next 3 years a 100% increase in investment in rare diseases. In Italy trials in this sector increased from 66authorised in 2010 to 167 in 2015 [3].
There are few medicines available for these diseases, just as in Italy the designation of an orphan drug is limited to the medicine that has to treat a very serious and/or disabling disease.
In this context, it is possible to identify different stages managed at different levels from the European level of market access to the national level of redeemability. Special national, European and international legislation has been adopted to reduce this divergence between supply and demand. The paths taken and the measures implemented have both supported research into rare diseases, through planning measures and economic incentives and at the same time provided criteria for the evaluation of cost-effectiveness.
The national health system has introduced measures such as price negotiation and reimbursement in balancing the economic interests of manufacturing enterprises with the right to health in this sector, through fairness of access to such medicines. Similarly, this article carries out a multidimensional analysis, capable of assessing progress in this area through research projects, of measuring economic conditions against the hoped-for clinical benefit, in order to ensure a balance between health and sustainability, through the regulation of both universalist health systems such as that of Italy and mutualists of the United States.
2. The Regulatory and Programming Framework
According to a historical reconstruction, the United States was among the first health systems to have
become aware of the need for regulation through the Orphan DrugAct1983, while others in Japan and
Australia, In the 1990s, the approach followed was to support production through economic aid measures,
such as incentives. The experience of the United States and Japan has shown that the most effective incentive
to invest in the production and marketing of such medicinal products is to ensure market exclusivity through
a patent to be extended for years necessary to enable the recovery of investment in research. The market
exclusivity concerns the therapeutic indication for which orphan medicinal product status is obtained,
without hindering the marketing of the equivalent also for rare disease patients.
The Regulatory Agencies of the Food and Drug Administration in America at the EMA in Europe and the AIFA in Italy have ensured the balance of health protection, with the economic interests of manufacturing companies and the sustainability of health systems both mutual and universalistic. In this regard, a coordinated effort can be identified at national and European level between industry and regulatory authorities (EMEA - European Agency for the Evaluation of Medicinal Products) by pursuing the dual objective of promoting and supporting the development of orphan medicinal products, ensuring their safety. With regard to the first profile, the direction taken has been to offer the necessary incentives for research and production and to manage in the most equitable way the system of reimbursement of the price charged to the Health Services. With regard to the second profile, the role of the Regulatory Agencies is to monitor the approval process of orphan drugs until they are placed on the market.
At European level, the lack of interest in orphan drugs in some states, such as Austria, was surpassed in 1998 by the start of a coordinated programming process. In France, since the 1990s, the production and access to such drugs has been facilitated by financing and tax exemption for manufacturing companies, from the centralisation of authorisations and the provision of expenditure ceilings for health facilities.
The objectives pursued by the European authorities are to support small and medium-sized enterprises in research and development of drugs or to stimulate these sectors and foster knowledge through communication and exchanges between research centers, institutions and patients. search for medicines for rare diseases, in 2000 Europe launched a specific program to designate an orphan drug, through measures such as scientific advice on study protocols, tax relief and exemptions and access to specific grants. Orphan medicinal products with qualification requirements qualify for commercial exclusivity in Europe for ten years, if there is another treatment available, the new drug will be considered a significant clinical benefit compared to existing options.
In view of the above, Ema’s Committee for Medicinal Products for Human Use recommended the approval of nine medicines in 2018, the Tegsedi, for patients suffering from hereditary transthyretin amyloidosis, able to influence the course of the disease and improve quality of life. This medicine is designated an “orphan” following an accelerated evaluation of the Ema, reserved for those of greatest interest, such as the Mylapta (metreleptin) designated an “orphan”, subject to a positive opinion in the treatment of leptin deficiency.
In Europe, the disease is rare when it affects no more than 5 people every 10,000 inhabitants [5].
At European level, actions aimed at promoting the production and distribution of orphan medicinal products were initially delayed because there was a lack of a single policy on orphan medicinal products, compared to the United States, for the fractionation and dispersion of competences in the field of health. Since 1995, Europe has laid the foundations for unified regulation, in order to promote a competitive market by preventing the dispersion of resources and removing legal and bureaucratic barriers to trade. The Community Regulations have defined the criteria for obtaining authorisation and identified the procedures for receiving incentives to promote the research, development and marketing of orphan medicinal products.
In 1999, the European Union adopted Reg. CE 141/2000 and 847/2000, which defined the criteria and procedure for the approval and designation of orphan drug by the Committee for Orphan Medicinal Product (COMP) of the European Medicines Agency- EMA and the way in which production incentives are awarded.
Reg. CE n. 141/2000on the initiative of France and the promotion of EURORDIS, was inspired by the regulation of the United States, in simplifying the authorization procedure, promoting research and distribution of orphan drugs in support of the pharmaceutical and biotechnology industries. To this end, it introduced a single centralised procedure for orphan status only for medicinal products intended for human use excluding veterinary medicines, medical devices, food additives and dietary products. A Committee for Orphan Medicinal Products (within the European Agency for the Evaluation of Medicinal Products (EMEA) is responsible for examining applications for designation, advise and assist the Commission in discussions on orphan medicinal products.
As a transparency measure, medicinal products designated as orphans are included in the Community Register of Orphan Drugs (COMP) [6].
Further measures have facilitated the production, through protocols of assistance for the pharmaceutical firms, (consultations and scientific opinions on test and clinical experimentations) in conformity to the anticipated requisite from the normative European. In addition, the European Union has encouraged the entry of new companies, such as sponsors qualified as such, who should benefit from the incentives granted by the Community or the Member States for the purpose of competition, capable of promoting research and development into medicinal products intended for the treatment of such diseases.
For the development and of the immission in commerce of the orphan medicines, the art. 8, c. 1 have expectation the exclusive one of market for ten years, in which you/they cannot be acetate question or granted authorizations to the immission in commerce, neither extensions, when they exist medicinal analogous, with the same therapeutic indications. The art. 9 Regs. Us 141/2000 bring that the orphan qualified medicines can profit of incentives made available from the Community and States members with the purpose to promote the search, the development and the immission of it in commerce and of measures of help to the search for the small ones and averages it undertook anticipated from the programs picture of technological research and development. The orphan medicines in Europe are to administer for pathology able to be in danger the life or weakening in chronic way; for a rare clinical condition, defined by a prevalence than no more than 5 subjects every 10 thousand individuals, calculated to level of the Union Europea;non you/they must be available valid treatments or, if the new medicine they are available you/he/she must represent a meaningful clinical benefit.
The authorization to the marketing (AIC) of an orphan medicine is released through the European procedure centralized near the EMA. In some cases, when for a specific medicine an marketing is necessary on in rapid times, before the necessary studies to the compilation of the dossier are completed, you/he/she can be granted from the EMA a positive opinion to the authorization with conditioned (under conditionalapproval), annual and renewable approval. The conditions of access to the procedure of conditioned authorization are the relationship positive benefit / risk; the indication of data complete clinicians; to answer to dissatisfied demands; benefits for the public health, consequential from the immediate availability of the medicine.
Completed the studies on the safety and effectiveness of the rare medicine and gotten the favorable opinion of the CHMP, they are not anticipated other obligations of the applicant the European authorization. The Reg. (CE) n. 847/2000 indicated the criteria for the designation of an orphan medicinal product, distinguishing them from the concepts of a similar or clinically superior medicinal product [7].
A marketing authorisation for a medicinal product may be granted in exceptional circumstances as specified in art. 14, c. 8, Reg. CE 726/04, in the treatment of very rare diseases, where the effectiveness and toxicity are not proven, with respect to which the applicant pharmaceutical company is not obliged to provide complete information. However, the holder of the AIC has to comply with the product safety obligations by managing specific controls. The confirmation of authorisationshall be linked to the annual review of those circumstances.
Article 17 of Recommendation (2009/C 151/02) on action in the field of rare diseases. e) plans to share Member States’ evaluation reports on the therapeutic or clinical added value of orphan medicinal products at Community level where relevant knowledge and expertise is collected, in order to reduce waiting times for access to orphan medicinal products for patients with rare diseases”. As a result of these regulatory actions, the European Medicines Agency has drawn up a list of rare diseases and provided for specific forms of access to these medicines, to which European countries have adapted. Subsequent evaluations of therapeutic value, price negotiation with pharmaceutical companies and exemptions remain the responsibility of individual countries. This division has increased the diversification of approaches in access, negotiation time, costs incurred by companies in research and production, in finding such drugs at affordable prices, in providing for forms of reimbursement (as in Austria).
The marketing approval does not imply the immediate availability of the drug, as it follows the steps in each country, in order to lay the conditions for the management and negotiation of the cost. Italian legislation protects the testing of orphan drugs and entry into the market to ensure patients have access to the best available therapies.
In Italy, a patient with a rare disease can access the orphan drug through the tools provided for in the
legislation in force. The National Health Plan 1998-2000 provided “protection of subjects affected by rare
diseases” to set up the national network, established by D. M. n. 279/2001, consisting of regional prevention,
surveillance, diagnosis and therapy, coordinated by the Ministry of Health. Another informative tool is
the national register for rare diseases, at the Higher Institute of Health, which collects the clinical and
epidemiological data of patients suffering from rare diseases according to all. 1 of the Decree. The administrative
jurisprudence has clarified, that despite the reform of Title V and the division of the legislative power in
matter of protection of the health, the norm referred to in the D.M. n. 279 is to apply to the regions, in order
to ensure the implementation of the organisational tools.
When the European authorisation for the placing on the market of an orphan drug is lacking, a patient with a rare disease may access such therapies through the procedures provided for in L. N. 648/19961, which allows the delivery of the S.S.N., in Deficiency of viable therapeutic alternative. For each drug if the Scientific Technical Commission at the AIFA expresses a positive opinion, it is included in an annex list, the method of use and the parameters subject to Pharmacovigilance.
In Italy the drugs for the therapy of rare diseases have different collocations, in the band C foreseen by L. 648/1996, band H at the patient’s expense, with difficulty of dispensation, finally no placement. As well as can be administered in a “compassionate” way, according to ministerial Decree 8 May 2003, with prescription on a nominal basis.
Access prior to marketing authorisation, during the third phase of clinical trials, is permitted when the degree of presumption of safety and efficacy is elevated. The actionable procedures is that the pharmaceutical industry that is developing a drug forwards a temporary authorisation to the administrative authority for a group of patients (ATU cohort in France and Italy, or authorization of compassionate use in Other European countries) is valid for a limited period of time in the country concerned. The other viable road is the medical request for temporary authorization to the administrative authorities, nominative valid for a specific patient and a limited period of time in the country concerned.
GERMANY - No Easy - Anything Special
AUSTRIA UC/NP – Slow- nothing particular
BELGIUM UC/NP -slow - nothing Particular
DENMARK UC/NP -Complex - nothing particular
FINLAND UC/NP – Complex - nothing particular
FRANCE- Rapid Coordination at the WHO level
SPAIN UC/NP - Classic -Nothing Special
GREECE UC/NP Classic -Nothing Special
IRELAND UC/NP- Classic - Nothing Special
ITALY YOUR - Classic - Nothing Special
LUXEMBOURG UC/NP – Classic - Nothing Special
NETHERLANDS UC/NP - Classic - improvement to be discussed
PORTUGAL depends on the casespecific funds assigned
UK UC/NP Slow considered onerous
SWEDEN UC/NP Easy Nothing special.
The exposed framework shows considerable diversification from the modalities of access to orphan drugs in the European States, this could foster mobility towards states where regulation makes administration not only simplified, but also more quickly, regardless of how you purchase and refund the price of the drug sold.
The above mentioned legislation provides for the distribution of the excess expenditure shelf to be distributed
among pharmaceutical companies competing for expenditure on direct purchases on the basis of market
shares. The previous regulatory approach provided for a breakdown of the budget for each farm on the basis
of the previous year’s expenditure and available incremental resources. The surplus expenditure generated by
orphan drugs, in addition to the funds provided for, was covered by other farms. In view of the uncertainty
of the forecast of these surpluses, potential distortions in planning were created for the holdings themselves.
The pay-back measure involves the participation of pharmaceutical companies in the cost of rare medicines. The system provided by the Budget Law of 2014 at the art. 1 co. 228 excluded from pay-back the turnover of orphan drugs “de facto”, without that qualification, because they were approved before the definition in the 1999 European Regulation.
The budget law 2019 simplifies the criterion of participation in payback, attributing to the’Aifa to determine the market share of each pharmaceutical company on the basis of turnover and compensation pro quota of exceeding the pharmaceutical spending ceiling (art. 1 co. 575 and 578).
L’art. 1 co. 575-584 Financial law 2019 simplifies the criterion of participation of industries in the pay-back, L’art. 1 co. 578-580 require Aifa to determine the market share of each pharmaceutical company on the basis of turnover and the compensation pro rata of the possible exceeding of the pharmaceutical expenditure ceiling. As a measure in support of medium-sized enterprises with a turnover of less than three million euro, they are exempt from participation in pay-back (art. 1 co. 579 lett. a), as an incentive for research and development for rare diseases.
The Financial law art. 1 co. 578 excluded the turnover calculations of the «medicinal products included in the Register of Orphan Medicinal Products for Human Use of the European Union», as provided for in the European Regulation, which defines the orphan medicinal product. However, this register does not include drugs “orphan-like” and those without market exclusivity.
The Financial Law 2019, without affecting the guarantees of treatment simplifies and makes more transparent the arrangements for participation in health expenditure, in order to prevent litigation. In accordance with the principle of progressivity, a fairer system will apply, which pre-determines the expenditure ceilings and excludes from the shelf the orphan drug invoices included in the list of the european medicines agency promoting research and control in production, with positive effects on the health system. The simulations of the applicationl. 2019 on 2017 data show that companies/groups with a cost value for direct purchases of upto 50 million will reduce the standard burden by 37,97%; for direct purchases upto 100 million the charge will be 20.39% and for those over 100 million the top charge is 112%”. The financial law 2019 allows companies producing orphan drugs to iniziate negotiation procedures with the process of submitting the dossier in the application for price and reimbursement and access to the accelerated negotiation procedure. In addition, innovative indicated orphan medicinal products are recognised as additional benefits such as direct access to regional treatment leaflets and economic incentives, in addition to the numerous incentives provided for by European legislation, including the 10-year market exclusivity to promote research and development. A simulation based on expenditure data for the year 2017 assesses the impact of the amendments provided for in the Budget Law 2019 on the side of the excess expenditure borne by orphan drug holders. The variation of the shelf to be imputed to the holders of medicines who contribute to the expenditure for direct purchases.
Budget law 2019 eliminates the criticality arising from the lists for orphan medicines, European and Italian. This simplification allows for greater transparency by eliminating the inequality according to which a medicinal product which is not on the EU list, because it is unfit or because of the choice of the owners of the medicinal products, but considered orphan in Italy, benefits of the economic advantage of being excluded from the excess of expenditure generated.
According to the 2019 financial regulation, medicinal products not on the EU List of Orphan Medicinal Products (Community Register of Medicinal Products designated as Orphans) cannot be considered orphan medicinal products in Italy.
3. Actions and Measures for Sustainability
In pursuing the objective of equitable access to pharmacological treatment, AIFA pays attention to patients
suffering from rare diseases, compared to this therapeutic need through the following paths. For the benefit
of patients, the normal and extraordinary procedures for access to the orphan hospital medicinal product or
of exceptional therapeutic and social importance, assessed as a priority by the extraordinary sessions of the
Commissions, shall be maintained, within 100 days of the request. Further facility for companies to apply
for classification and price prior to granting marketing authorisation remains confirmed.
Rare diseases have a threshold of no more than five out of 10000 patients, according to Reg. (EC) No 141/2000 European Parliament and Council, of 16 December 1999, concerning orphan medicinal products. 10. These are serious, chronic and potentially lethal diseases. There is no common international list of rare diseases, but lists can be found on sites of the National Organization for Rare Disorders (NORD) and the Ministry of Health. The d. M. 279/2001, Reg. establishment of the national network of rare diseases and exemption from participating in the cost of related health benefits) provides for a list of more than 500 rare diseases whose benefits are exempt.
The AIFA for the protection of health promotes tools to promote the economic balance of the national
health system, through the negotiation of prices and the vigilance not only of the effects of orphan drugs,
but also the observance of programmed spending limits.
In accelerating the availability of orphan drugs, the Balduzzi Law (Law 189/2012, art. 12, c.3) established that the pharmaceutical company that owns AIC of orphan drug may submit a price and reimbursement request to AIFA after the positive opinion of the CHMP. The c. 5-bis of the same article (inserted by the art.44 for the classification of orphan drugs and of exceptional therapeutic relevance, of the DL n. 69/2013, converted into L. n. 98/2013), states that the AIFA evaluates as a priority this classification for the purpose of reimbursement by the NHS. Orphan drugs of exceptional therapeutic relevance for which the related application was submitted with documentation. In this case, the deadline for evaluating is reduced to one hundred days (“fast track authorization”). In case of no-show within thirty days of the granting of an orphan drug of exceptional therapeutic importance, AIFA requests the company that is the holder of the relevant marketing authorization to submit the classification application and refundable within thirty days.
AIFA is the first agency in Europe to promote independent research on orphan drugs for public and nonprofit institutions. This originates in the recognition, the subject of scientific debate, of the role of independent research in areas of impact for health, but with an insufficient commercial interest. As a financing measure, the law establishing the AIFA has set up a fund for the use of orphan drugs for rare diseases pursuant to art. 48, c. 19, lett. a, D.L. n. 269/2003, in L.n. 326/2003. This fund is fueled by 5% of annual expenses for the promotion activities of pharmaceutical companies. The use of the fund is dedicated for the 50% to the purchase of orphan drugs for rare diseases that represent a hope of cure for serious pathologies; and the remaining 50% in research on the use of such drugs (comparative clinical studies between drugs aimed at demonstrating additional therapeutic value and appropriateness). With regard to the purchase of these drugs, the requests for access to the fund are forwarded to AIFA, through the Regions, the Reference Centers that treat the sick, or from specialized structures, with the definition of the diagnosis and the plan therapeutic.The requests coming from the single Centers aware of the Region can be accepted by the AIFA, through a documentation necessary for requesting access to the fund, any supporting scientific literature, a clinical report with therapeutic plan for each patient. The funding request is supported by the dosage per cycle, number of cycles and unit cost of the medicine. The request is evaluated by the Scientific and Technical Advisory Committee of the AIFA which expresses its opinion, subject to verification of the conditions required by law.
This originates in the recognition, the subject of scientific debate, of the role of independent research in areas of impact on health, but with insufficient commercial interest. The projects financed by AIFA in Independent Research aim to create scientific evidence and guarantee adequate repercussions of the scientific results. The AIFA has implemented these objectives between 2005-2007, with calls for proposals dedicated to orphan drugs. In 2008, the Ministry of Health with the ban on rare diseases funded € 3,000,000 for 12 studies. In improving the care of this group of patients, the studies assessed the benefit / risk of orphan drugs for rare diseases, approved or designated by the EMA and in off-label treatments in rare diseases. In the call for proposals concluded in 2016, 31,294,724.05 have been allocated for 23 projects related to “rare diseases” for about 20 million and 7 involve the study of the use of orphan drugs for an amount allocated of 5,022,954 euros. Another call is to be launched within the first quarter of 2019.
In the call for proposals concluded in 2016, 31,294,724.05 have been allocated for 23 projects related to “rare diseases” for about 20 million and 7 involve the study of the use of orphan drugs for a portion of € 5,022,954. Launch of the AIFA 2018 call for tenders: 6.5 million euros for independent research which provides for the allocation of 6.5 million euros to finance research projects conducted within the thematic areas identified by the Agency, including rare diseases.
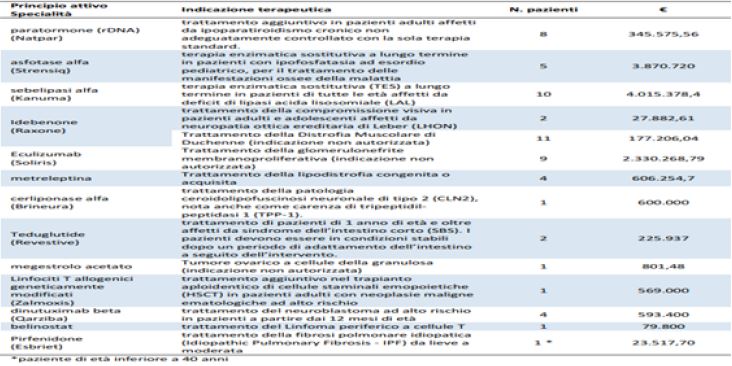
Recently AIFA has initiated a systematic process of involving patients and representative associations in order to extend participation through involvement in all stages of the decision-making process.
Another way of administering the orphan drug is the off-label in the treatment of rare diseases, for which it
can be the only possible therapy. Very severe, no treatments are available, or when existing ones have proved
ineffective.
In an article in the Journal of Pharmacology and Pharmacotherapeutics a debate emerged about the efficacy and safety of off-label use for rare patients. On the one hand, the need for a greater role of the regulatory authorities in guiding these therapies is highlighted, on the other hand the benefits of the off label are highlighted, for patients, doctors and pharmaceutical companies, since treatment would be allowed, even if not approved, but whose safety has been ascertained.
Indeed, in France and the United States the regulation has implemented the risk-benefit analysis, in ensuring greater appropriateness in the off-label use of rare drugs and better information for users. a research group from the University Hospitals Leuven interviewed seven specialized doctors and six experts on health policy related to orphan drugs in 2013. From this study it emerged that such off label treatments require certain guarantees such as the indication expressed in the illustrative leaflet is general. Furthermore, this strategy should only be considered in cases where, for a very serious condition, specific therapeutic options have proved ineffective or are not available at all. Some of the subjects interviewed argue that off-label use of drugs, particularly orphan drugs, should be used only in clinical trial regimens, or that it should be performed only in specialized hospital centers [8].
In the preamble of the decree the Minister of Health of 11 February 1997 and the opinion of the Superior Council of Health highlights the need for adequate regulation also of the therapeutic use of medicines not yet approved and subjected to advanced clinical trials on the Italian territory or in foreign countries.
In Italy for orphan and off-label drugs, the regulatory reference is made up of Law n. 648/1996 followed by the provision of 20 July 2000 in which a list of medicinal products that can be supplied is established, in the absence of a valid therapeutic alternative, charged to the National Health Service. The pharmaceutical company can request to register a drug as an orphan for a certain rare disease, in order to speed up registration and extend the fields of use of the off-label drug through further research and experimentation. Off-label medicines can be included in such special list not only on the initiative of the Commission, but also through the proposal of patient associations, scientific societies, public or private health bodies. The medicines included in the list can be prescribed by specialized hospital or university facilities or by scientific and hospitalization institutes.
The off-label, in this context, is to be implemented in guaranteeing the possibility of treatment, with the same level of effectiveness as other drugs. However, the off label prescription requires the health care special attention in supervising the side effects or any ineffectiveness, compared to the expected expectations.
The use of off-label drugs must be supported by experimental tests and / or scientific documents that can support the choice made if it refers to the possible advantages / disadvantages of consolidated treatments for the indication. The element that favors the choice of off-label use of a drug is the lack of valid therapeutic alternatives, ie no treatments are available, or when existing ones have proved ineffective.
The dynamics of prescription and administration of the off-label are different [10]. The administration of the off-label is reimbursable within the NHS, under certain conditions, when the orphan drug is less expensive, if used outside of therapeutic indications of administration. This is possible in a defined scientific context, if a valid therapeutic alternative is not available, as every economic evaluation withdraws respect for the protection of the right to health for rare diseases.
The dispensation of these medicines, payable by the health system, can be carried out by the pharmaceutical service of the prepared structures, if the drugs are available in local pharmacies or by the pharmaceutical service of the local health authorities of the patients’ residence. In this last situation, the expense is borne by the local health care company of residence. The dispensation of these medicines, payable by the national health system, can be carried out by the pharmaceutical service of the prepared structures, if the drugs are available in local pharmacies or by the pharmaceutical service of the local health authorities of patients’ residence. In this situation, the expense is borne by the local healthcare company of residence.
In conclusion, the off-label use of orphan drugs can be of real benefit in the treatment of rare diseases, based on medical justifications, clinical evidence and regulated by regulations and guidelines that protect interest and safety of patients.
4. Conclusions: The Social Welfare Impact, Comparison Between Economic and Organizational Measures and Balancing Instruments
This concluding paragraph will analyze the impact of orphan drugs on the national health system, through the setting of the welfare economy. The latter assesses possible situations, including alternatives according to the criteria of efficiency and equity, in order to compare the results deriving from the action of economic operators without public intervention with those obtained when the State intervenes.
The increasing attention paid to diseases called “rare” has not allowed to overcome areas “forgotten” by pharmacological research and production, maintaining access inequalities for groups of users. These issues need to analyze the organization of health systems where measures are intertwined “push ”and“ pull ”, in supporting research and production but also in ensuring the safety of these drugs to protect the health of the rare patient. In order to overcome the criticalities of the rare drug market, some states have launched solidarity actions, a commitment to purchase a share of these drugs and thus guarantee a demand share to the producers. This set of measures can also determine a conflict between the expenditure containment objectives with respect to the fairness of access to these drugs.
Another objective is the transparency of the supply chain of these health products, through measures aimed at reducing information asymmetries through greater dissemination of information and the implementation of participation. The growing synergy between doctor-patient-companies has supported the professional activity of individual professional operators, but this was not enough. The balance of pharmaceutical expenditure implies not only an assessment of the possible burdens of the national health fund in reimbursing the cheaper drug, but also choices in the allocation of available resources. Each of the interested parties has a role and as such it is necessary to guarantee the widest participation in order to guide the final choices through their own needs.
On the one hand, the costs incurred by the companies in the research, development, production, marketing and distribution of these drugs are financial costs of Goods Sold, which are subtracted from the sale price. Considering these costs would allow a fairer reimbursement price to be determined in guaranteeing greater access to these drugs and better allocation of public and private resources to support this sector of research and pharmacological production.
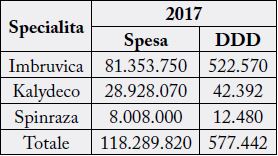
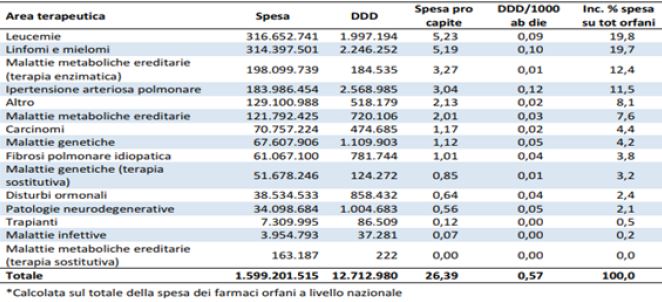
The evaluation of the value offered by the asset, based on the “marginalist Cost x QUALY and“ willingness to pay ”method is to be contextualised for orphan drugs with respect to the growing demand and the constraints of economic sustainability. The achievement of the optimal condition implies to implement the negotiation tools with the regulatory authorities, such as the AIFA, in defining the price and the repayment quota. The achievement of the balance between the supply and the availability of user spending and those who reimburse, requires the necessary state intervention in balancing the economic interests of each economic operator involved in the production and distribution processes, with the therapeutic ones of health professionals and users involved in the assistance circuit and public ones oriented to the sustainability of the national health system.
The optimal condition implies to define the price and reimbursement object of negotiation with the regulatory authorities, such as AIFA, identifying the intersection point of the curves between the offer of the seller (Business) and the availability of expenditure of those who buy which user and those who reimburse, able to guarantee a balance in the national health system.
In order to overcome the persistent shortage of medicines, the Health Minister has convened a table with the institutions and actors involved in the production and distribution of medicines, in order to arrive through shared choices to overcome the critical issues exposed.
Bibliography


Hi!
We're here to answer your questions!
Send us a message via Whatsapp, and we'll reply the moment we're available!